Targeted Therapeutics
Wednesday, July 12, 2023
PROGRESS ON TARGETED THERAPEUTICS: PERSONALIZED MEDICINE AT MARY CROWLEY
“JAMA Oncology reports statistically significant advantages in response rates between personalized and non-personalized therapies.”
A primary strategy in cancer treatment development is to construct a therapeutic with the ability to:
1.) differentially target cancer cells rather than normal cells;
2.) have a companion biomarker able to identify which of the patients diagnosed with a cancer have the relevant and accessible target;
3.) be cytotoxic (i.e., kill cells) or, more likely, contribute to cytotoxicity in combination with other targeted therapeutics;
4.) be systemically administered (to reach both primary and metastatic cancer sites);
5.) penetrate deeply into tumors containing multiple billions of cancer cells;
6.) be homogeneously (i.e., evenly) distributed throughout the tumor volume; and
7.) be available to all candidate patients within the constraints of medical economics.
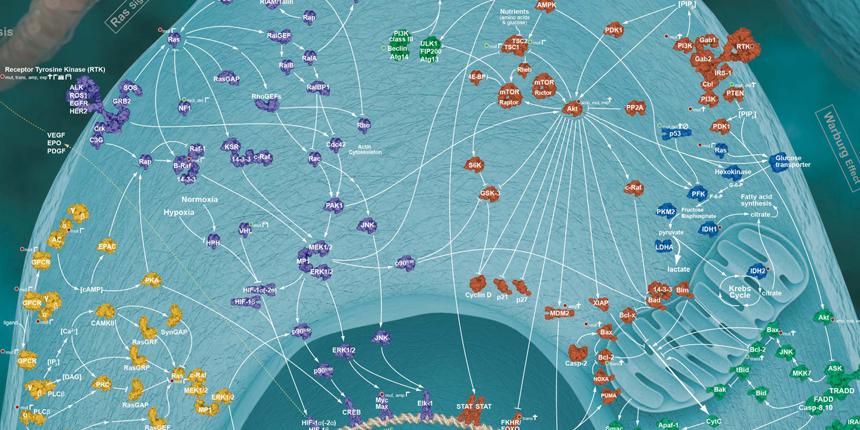
The traditional pillars of cancer therapy—surgery, radiation, and chemotherapy—are tactical approaches limited in scope. Both surgery and radiation, to some extent, geographically target cancer but are spatially localized and regionally limited by surrounding normal tissue damage, which can result in adverse side effects. Chemotherapy, albeit systemically administered, attacks processes essential to cell growth and division and are non-specific. Chemotherapy discriminates between cancer and normal cells on the basis of quantitative differences in growth kinetics and lack of DNA repair fidelity—thus, a relative specificity with a narrow “therapeutic window”, i.e., the difference between the concentration needed to affect cancer cells and that which produces adverse side effects. Targeted cancer therapies, on the other hand, capitalize on the quantitative and, more importantly, qualitative differences resulting from the genetic (mutation or copy number alterations) and/or epigenetic (non-structural genetic modifications) changes that rewire the biomolecular cellular network creating new pathways with distinctive fragile gene/protein nodes (see figure 1).
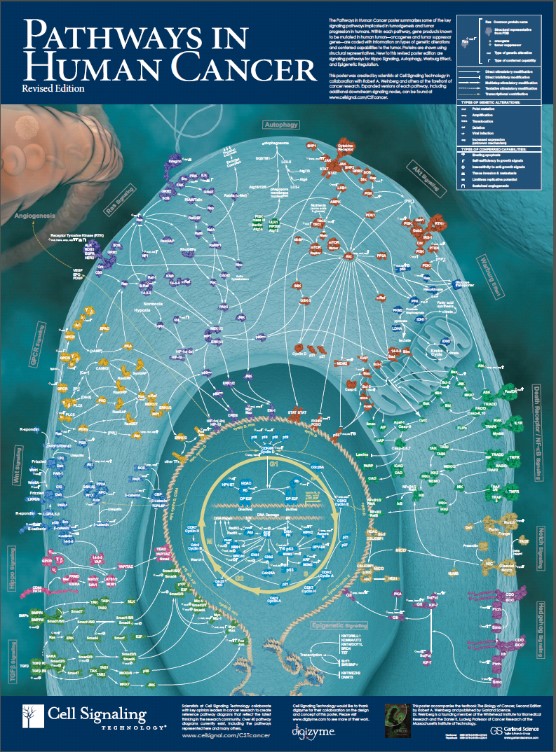
Figure 1: Rewired cancer-signaling network comprising gene/protein nodes and interconnected linkages.
Despite a bewildering array of potential targets, including mutated and epigenetically modified genes, copy number changes, mRNA splicing differences, and post-translational protein modifications, the vast number of nodes can be classified into one or more of 12 major pathways, the interaction of which confer an adaptation advantage to the evolving cancer (see figure 2). As a result, the goal is to identify the dominant pathway[s] amongst the 12 in an individual cancer and target the rate-limiting nodes therein.
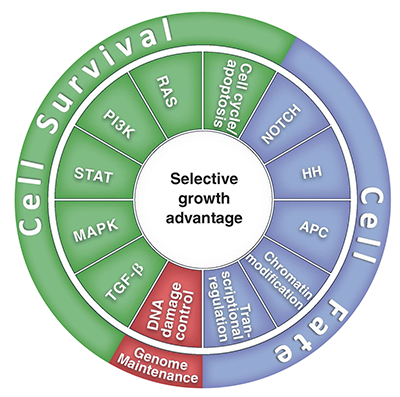
Figure 2: Cancer cell signaling pathways and consequent processes.
FDA-approved targeted therapies are classified as either monoclonal antibodies (MAb) or small molecules. The MAbs target specific antigens on the cancer cell surface, generally transmembrane receptors that initiate a signaling cascade. The small molecules can interact with proteins inside the cancer cell able to interfere with enzymatic activity. Thus far, all of the approved small molecules are directed against kinases (an enzyme that adds a phosphate group (-PO4) to a protein to modulate its function).
In a recent meta-analysis (JAMA Oncol. doi:10.1001/jamaoncol.2016.2129) of 346 Phase I studies involving 13,203 patients, 272 of the studies were performed in patients with solid tumors. A statistically significant advantage in response rate (RR) was found for personalized versus non-personalized therapy, 24.5% versus 4.5% (p<0.001), respectively. Further, the median RRs were higher for targeted genomic alterations than for protein aberrations (overexpression), 42% versus 22.4% (p=0.001), respectively. In another meta-analysis (J Clin Oncol. 2015. 33:3817-25), 570 Phase II studies comprising 32,149 patients were analyzed. As a result of more extensive protocol endpoint data, comparing personalized to non-personalized targeted therapy, statistically significant improvements were found in RR (31% versus 10.5%; p<0.001), progression-free survival (PFS: 5.9 versus 2.7 months; p<0.001), and overall survival (OS: 13.7 versus 8.9 months; p<0.001), respectively. Similar to the Phase I study analysis, personalized therapies chosen on the basis of genomic rather than protein biomarkers achieved statistically significant improved endpoints (RR: 34.1% versus 24.2%, p=0.027; PFS: 6.4 months versus 5.0 months, p=0.015; OS: 16 months versus 8.2 months, p=0.007).
Patients and oncologists at Mary Crowley Cancer Research have significantly contributed to the development and final FDA approval of a wide variety of personalized cancer therapeutics with 1,629 patients treated with small molecules and 192 patients with MAb in Phase I and II trials through 2014 via coordinated efforts with strategic partners (see table 1).
| Strategic Partners | Mary Crowley Cancer Research |
| Genome analysis -> potential target | Bioinformatics integration |
| Community practice participation | Rapid data turn-around time |
| Update clinically relevant signals | Rapid relevant signal addition |
| Development of protein based signal detection | “-omic” integration to facilitate pathway ID |
| Capacity to re evaluate new signal diagnostics rapidly with patient samples online | Large patient signal specific cohorts for trial testing |
| Understanding of common utilized signals for experimental therapeutic testing | Understanding of potential toxic relationship to signals |
| Information/publication signal expression to patient benefit | Cooperative meeting presentation |
| Companion diagnostic opportunity with biotech | MCCR custom report |
| Cooperative meeting presentation | Cooperative site education |
| Cooperative site education | Simplification planning of relevant signals |
Table 1
Given the hypothesized advantage of targeting therapeutics to genomic rather than protein nodes, Mary Crowley, in collaboration with Strike Bio, Inc., has embarked on a challenging new project. Almost all approved drugs currently on the market are directed towards protein targets. The druggable proteome is defined as the fraction of proteins that have the ability to bind a small molecule or antibody with the required affinity and the appropriate chemical properties. Of the 22,000 protein coding genes, approximately 400 (2%) have recurrent mutations in cancer, of which only 80-200 are druggable. However, given the technical development of next generation sequencing (NGS) of the genome (DNA) and transcriptosome (RNA), Mary Crowley and Strike Bio, Inc. are spearheading the application of an innovative strategy, RNA interference, to block the expression of mutated cancer driver genes that are otherwise non-druggable. Activating K-Ras mutations (see figure 2) are detected in 20% of all cancers and 90% of pancreatic adenocarcinomas. In order to block protein expression from this oncogene, Strike Bio, Inc. has developed a therapeutic lipoplex (a DNA-containing liposome) consisting of a fusogenic liposome delivery vehicle with relative tumor selective properties and a plasmid encoding the RNA interference bi-shRNAK-Ras that both degrades K-Ras mRNA and blocks protein translation. The bi-shRNAK-Ras is a modular construct with three therapeutic warheads targeting the three most frequently occurring K-Ras mutations in pancreatic adenocarcinoma, G12D, G12V, and G12R. Pre-clinical studies in a K-RasG12D pancreatic cancer xenograft (an animal model) confirm bi-shRNAK-Ras safety, specificity, and effectiveness. This novel targeted therapy can be administered systemically, target a specified mutated gene product, and multiplex for multiple target combinations. The bi-shRNA technology is poised for clinic entry in a protocol targeting the Ewing’s sarcoma EWS-FLI1 fusion gene.

Related Posts
Discover more posts like this.